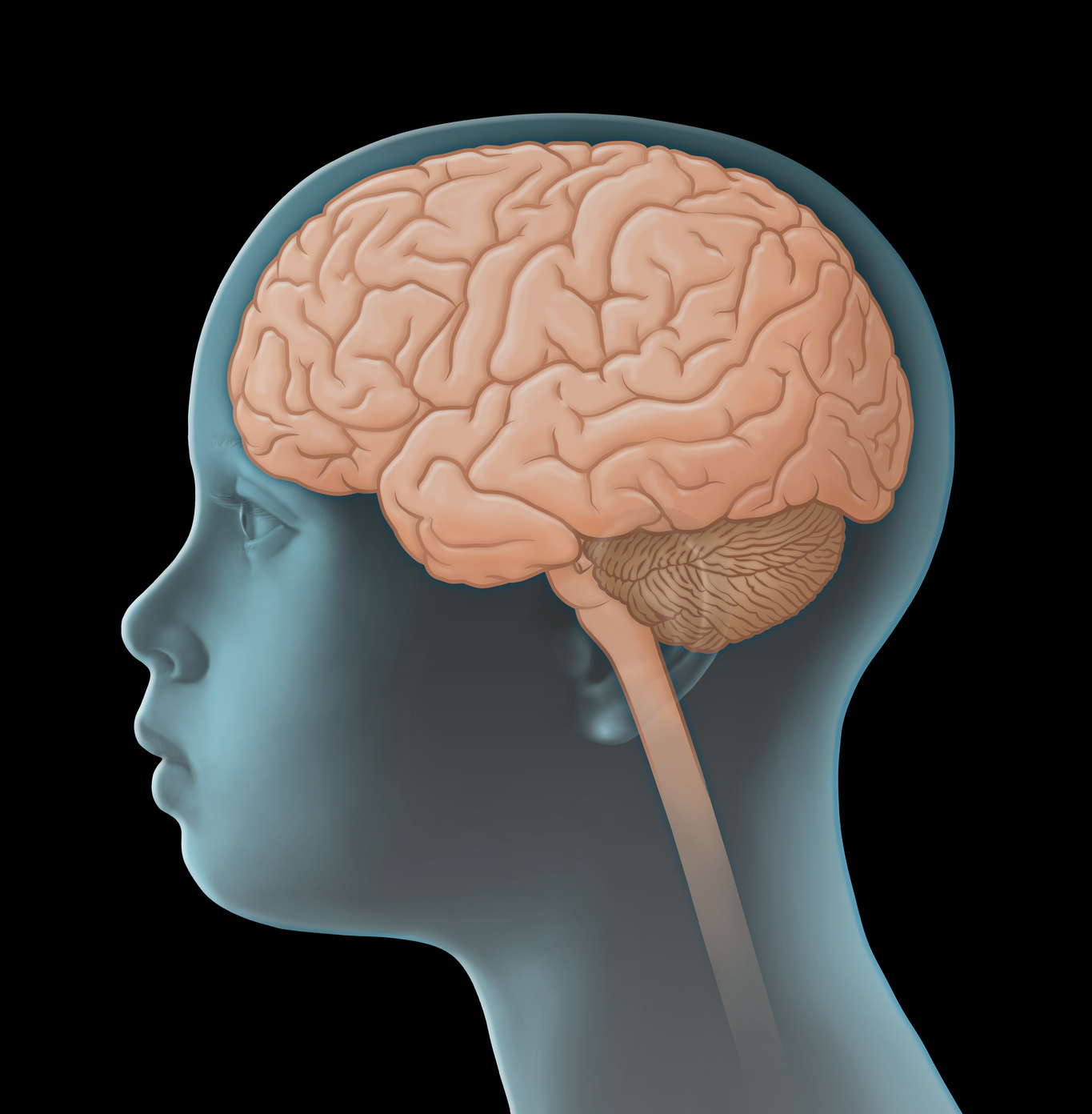
Merkezi sinir sisteminin rabdoid tümörleri (AT/RT) – Kısa Bilgiler
Rabdoid tümörler, diğer yerlerin yanı sıra merkezi sinir sisteminde de gelişebilen çok agresif büyüyen embriyonal tümörlerdir ve daha sonra AT/RT olarak adlandırılırlar. Aşağıdaki yazıda hastalık tablosu, olası hastalık sebepleri ve bulguları, hastalık seyri, tanısı, tedavi planlaması, tedavisi, tedavi iyileştirme çalışmaları, rehabilitasyonu, tedavi sonrası izlemi ve hastalığın prognozu hakkında detaylı bilgiler bulacaksınız.
yazar: Maria Yiallouros, Yayın İzni: Prof. Dr. Dr. med. Michael C.Frühwald, türk tercüman: Dr. med. Ebru Saribeyoglu, Last modification: 2025/06/03 https://kinderkrebsinfo.de/doi/e216137
Table of contents
Hastalık tablosu
Rabdoid tümörler nadir görülen, çok agresif büyüyen tümörlerdir ve çoğunlukla yaşamın ilk iki yılında bebeklerde ve küçük çocuklarda görülürler. Embriyonal [embriyonal] tümörlerdir, yani son derece olgunlaşmamış (ayrışmamış) hücrelerden kaynaklanırlar. Rabdoid tümörler vücudun tüm dokularında gelişebilir. Ancak en sık beyin ve omurilik (yani merkezi sinir sistemi) etkilenir (yaklaşık %65). Rabdoid tümörler ayrıca öncelikle böbreklerde ve karaciğerde ve yumuşak dokuda (örneğin boyun, uyluk ve göğüs duvarında) görülür.
Merkezi sinir sisteminin (MSS) rabdoid tümörleri "atipik, teratoid, rabdoid tümörler" (kısaca AT/RT) olarak da adlandırılır. Dünya Sağlık Örgütü'nün (DSÖ, WHO) MSS tümörleri için geçerli sınıflandırmasına göre (DSÖ sınıflaması CNS5) bunlar 4. derece tümörler olarak sınıflandırılır (toplam 4 dereceden). Bu nedenle özellikle hızlı büyüyen kötü huylu MSS tümörleri olarak kabul edilirler.
Aşağıdaki bilgiler sadece merkezi sinir sisteminin rabdoid tümörleri ile ilgilidir.
Rastlanma sıklığı
Merkezi sinir sisteminin rabdoid tümörleri (AT/RT) genel olarak nadir görülür. Almanya'da 18 yaş altı çocuk ve gençlerde görülen tüm malign hastalıkların sadece %0,6'sını oluştururlar. Bu, 1 milyon çocuk başına yaklaşık bir yeni vaka gibi ortalama bir sıklığa (insidans) karşılık gelmektedir. Avrupa Rabdoid Tümörler Kayıt Merkezine (EU-RHAB) göre, Almanya'da her yıl yaklaşık 15-22 AT/RT hastası kaydedilmektedir (EU-RHAB hakkında daha fazla bilgi için "Tedavi iyileştirme araştırmaları ve veri bankası" bölümüne bakınız).
AT/RT neredeyse tüm yaş gruplarında görülür, ancak yaşamın ilk iki yılındaki bebekler ve küçük çocuklar en sık etkilenenlerdir (yaklaşık %80 oranında). Alman Çocukluk Çağı Kanser Kayıt Merkezi (Mainz) verilerine göre, AT/RT 1 yaşın altındaki çocuklarda en sık görülen MSS tümörleridir. Altı yaşından itibaren hastalık sadece nadiren tanımlanmaktadır. Hastaların tanı anındaki ortalama yaşı yaklaşık 1,5 yıldır. Erkek çocuklarda kızlara göre biraz daha sık görülür (cinsiyet oranı: 1,3:1).
Yerleşimi ve dağılımı
EU-RHAB kayıtlarından elde edilen rakamlara göre (2005-2024 dönemi), AT/RT vakaların yaklaşık %50'sinde beyincik (serebellum) veya beyin sapı bölgesinde (yani infratentoryal) beyincik köprücük açısı (serebellopontin açı) olarak adlandırılan bölgede gelişmekte ve buradan çevre yapıları invaziv olarak etkilemektedir. Tümörlerin yaklaşık %34'ü beyincik zarı (serebellar çadırın) üzerinde (supratentoryal), örneğin büyük beyin (serebrumda) büyür. Geri kalan AT/RT epifiz bezi, ara beyin (diensefalon), omurilik bölgesine dağılır veya vücudun çeşitli yerlerinde görülür, yani multifokaldir (yaklaşık %2). Büyüklükleri nedeniyle çıkış yerinin tam olarak belirlenemediği tümörler de vardır. Yaşlı hastalarda supratentoryal rabdoid tümörler infratentoryal olanlara kıyasla daha sık görülür.
AT/RT'li hastaların %20–30'unda tanı anında, ağırlıklı olarak merkezi sinir sistemi içinde olmak üzere metastazlar mevcut olabilir. Öte yandan, MSS dışında metastaz son derece nadirdir ve neredeyse sadece bu tümörlere yatkınlığı olan çocuklarda görülür (rabdoid tümör yatkınlığı sendromu olarak adlandırılır). Vakaların yaklaşık %10-15'inde bu sendroma sahip hastalarda aynı zamanda vücudun diğer bölgelerinde de rabdoid tümörler gelişir (multifokal, senkron tümör istilası). Bu bir metastaz vakası değildir.
Sebepleri
Bir rabdoid tümörün gelişim nedenleri henüz tam olarak anlaşılamamıştır. Bununla birlikte, rabdoid tümörlerin neredeyse tamamının (yani %90 –95'inden fazlasının) – vücuttaki konumlarından bağımsız olarak – kromozom 22 üzerindeki belirli bir gende yapısal (epigenetik) bir değişikliğe sahip olduğu bilinmektedir. Bu gen, SMARCB1/INI1 proteininin üretiminden sorumlu olan SMARCB1 genidir (INI1 olarak da bilinir). Diğer şeylerin yanı sıra, bu protein hücre büyümesi ve olgunlaşması (farklılaşma) gibi hücresel mekanizmalarda önemli bir rol oynar. Genetik kusur (mutasyon), protein üretiminde başarısızlıkla sonuçlanır ve bu da hücre dejenerasyonuna ve tümör gelişimine yol açabilir.
Çoğu vakada, değişmiş SMARCB1 geni, somatik bir hücrede kendiliğinden oluşan dejenerasyon nedeniyle yalnızca tümör hücrelerinin kendisinde tespit edilebilir. Daha nadir vakalarda (tüm hastaların %25–30'unda), germ hattı (germline) hücreleri (germinal hücreler) ve dolayısıyla vücudun tüm hücreleri de etkilenir (germ hattı mutasyonu). Bu, embriyonik gelişim sırasında hastanın germ hattında kendiliğinden oluşan genetik bir değişiklikten veya (çok nadiren) ebeveynlerden birinden kalıtsal olarak geçen bir bozukluktan kaynaklanabilir. Her iki durumda da hastalık kalıtsaldır, yani değişmiş gen ve dolayısıyla rabdoid tümör geliştirme yatkınlığı yavrulara aktarılabilir. Uzmanlar bunu rabdoid tümör yatkınlık sendromu (ingilizce: Rhabdoid Tumour Predisposition Syndrome, RTPS) olarak adlandırmaktadır. Ancak SMARCB1 mutasyonu olan tüm hastalarda rabdoid tümör gelişmez.
SMARCB1 mutasyonlarına ek olarak, kromozom 19 üzerindeki SMARCA4 genindeki (aynı zamanda BRG1) mutasyonlar çok nadiren tümör hastalığının nedenidir. Bu mutasyon genellikle bir ebeveynden kalıtılır. Mutasyonun türüne bağlı olarak (SMARCB1 veya SMARCA4) RTPS1 veya RTPS2 olarak adlandırılır.
Bilmekte fayda var: Bir rabdoid tümör yatkınlık sendromu (RTPS1 veya 2), yani bir germ hatti mutasyonu mevcutsa, hastalığı olan bir çocuğun kardeşlerinde de hastalık riskinin artması muhtemeldir. Bunun nedeni, sendromun her iki formunun da otozomal dominant olarak kalıtılmasıdır; bu da RTPS'li bir ebeveynin çocuklarının da ailesel germ hattı mutasyonu taşıyıcısı olma olasılığının %50 olduğu anlamına gelir. Bu nedenle, kalıtsal bir rabdoid tümörden şüpheleniliyorsa, etkilenen hastanın ailesi için önleyici tedbirler önerilir (bkz. Bölüm "Tanı").
Hastalık belirtileri
Çok hızlı büyüme eğilimleri nedeni ile, çocuk ve gençlerde rabdoid tümörlere bağlı hastalık bulguları (semptomları), bir kaç hafta içinde ortaya çıkar. Diğer merkezi sinir sistemi tümörlerinde [MSS tümörü] olduğu gibi, merkezi sinir sistemi rabdoid tümörlerinin (AT/RT) sebep olduğu hastalık belirtileri, hastanın yaşına, tümörün merkezi sinir sistemi içinde bulunduğu yere ve yayılma derecesine bağlı olarak değişir. Spesifik olmayan (genel) ve spesifik olan (lokal) hastalık belirtileri ortaya çıkabilir.
Spesifik olmayan semptomlar (belirtiler)
Spesifik olmayan genel semptomlar tümörün yerleşim yerinden bağımsızdırlar ve merkezi sinir sistemi tümörü dışında başka hastalıklarda da görülebilen bulgulardır. Etkilenen ana grup olan bebeklerde ve küçük çocuklarda, örneğin tiz ağlama, huzursuzluk, yorgunluk, uyuşukluk, içme isteksizliği / yemeyi reddetme, kusma, gelişim ve/veya büyüme bozuklukları ile kendini gösterir. Sıklıkla kafatası sinirlerinin felci nedeniyle başın eğik durması da söz konusudur. Daha büyük çocuklar örneğin baş ve/veya sırt ağrısı veya baş dönmesinden şikayet edebilirler; performans sorunları, konsantrasyon sorunları ve karakter değişiklikleri de bir MSS tümörünün olası genel belirtileridir.
Bu semptomların sebebi kafatasının içinde yavaşça gelişen basınç artışıdır. Büyüyen tümör kitlesi kafa içi basıncı direkt arttırabileceği gibi tümör basısına bağlı olarak gelişen beyin omurilik sıvısının dolaşım bozukluğu da basınç artışına sebep olabilir. Beyin omurilik sıvısının dolaşımın bozulması hidrosefaliye de neden olur (beyin omurilik sıvısının kafa içinde birikmesi). Bu durumda bıngıldakları (fontanel) açık bebek ve küçük çocuklarda baş çevresinde hızlı bir artış meydana gelir (makrosefali).
Spesifik semptomlar (belirtiler)
Lokal (spesifik) semptomlar tümörün merkezi sinir sistemi içindeki yeri ve hangi işlevsel bölgeleri etkilediği konusunda bilgi verebilirler. Örneğin, beyincikteki bir rabdoid tümör hareket bozukluklarına, görme bozukluklarına, denge ve yürüme bozukluklarına neden olabilir; büyük beyindeki bir tümöre hemipleji, nöbetler (sara nöbeti) konuşma ve davranış bozukluklarının yanı sıra duyusal bozukluklar da eşlik edebilir. Omurilik bölgesindeki bir rabdoid tümör ise şiddetli ağrı ve çeşitli felç türleriyle (örneğin mesane-bağırsak fonksiyonu) dikkat çekebilir.
Bilinmesinde yarar var: bu hastalık bulgularının birinin veya bir çoğunun bir hastada bulunması, hastada mutlaka rabdoid tümör veya başka bir beyin tümörü olduğu anlamına gelmez. Yukarıda sayılan bulguların çoğu, beyin tümörü ile hiç ilgisi olmayan bir çok zararsız hastalıkta da ortaya çıkabilir. Adı geçen bulguların varlığında (örneğin sık tekrarlayan baş ağrılarında, küçük çocuklada orantısız olarak hızla büyüyen kafaçevresi olması durumunda), sebebi belirlenmesi için bir doktora danışmakta yarar vardır. Eğer gerçekten rabdoid tümör veya başka bir beyin tümörü söz konusu ise, çok hızlı bir şekilde tedaviye başlamak gerekir.
Tanı
Doktor/çocuk doktoru muayene sırasında hastanın öyküsünde (anamnez) ve fiziksel muayenesinde merkezi sinir sistemine ait kötü huylu tümör olabileceğine dair veriler elde ederse, hastayı özellikle çocuk ve gençlerde kanser ve kan hastalıkları uzmanı bir kliniğe (Pediatrik Onkoloji ve Hematoloji Kliniğine) sevk edecektir. Çünkü böyle bir tümör şüphesi durumunda hastanın kötü huylu MSS tümörüne sahip olup olmadığının anlaşılabilmesi için değişik alanlardaki uzmanların birlikte çalışmaları ve geniş kapsamlı tetkiklerle bir sonuca varılması gerekmektedir. Hastanın en iyi sonucu verecek, en uygun tedaviyi alabilmesi için gliomun tipinin belirlenmesi ve vücuttaki yayılma evresinin saptanması da gereklidir.
Tümör tanısı için kullanılan görüntüleme yöntemleri
MSS'nin rabdoid tümörü (AT/RT) şüphesi, dikkatli bir tıbbi öykü (anamnez), fiziksel ve nörolojik muayenenin ardından ultrasonografi ve manyetik rezonans tomografisi (MRI) gibi görüntüleme yöntemleri kullanılarak netlike kazanır. Hızlı bir açıklığa kavuşturmak için bilgisayarli tomografi taraması (acil BT) da gerekli olabilir. Bu yöntem ile merkezi sinir sisteminde bir tümör olup olmadığı anlaşılmış olur. Tümörün yeri ve boyutu ile komşu yapılarla olan sınırları da çok net bir şekilde görülebilir.
Doku örneği alınması (biyopsi) ve incelenmesi
Tanıyı kesin olarak doğrulamak için bir doku örneği alınmalıdır (biyopsi). Bu genellikle cerrahi bir işlem (operasyon) gerektirir. Alınan doku daha sonra histolojik, immünohistokimyasal, sitogenetik ve moleküler olarak analiz edilir. Özellikle değişmiş bir SMARCB1 veya SMARCA4 geninin saptanması rabdoid tümör tanısını kolaylaştırır (bkz. “Sebepleri” bölümü). İmmünohistokimyasal incelemenin bir parçası olarak, SMARCB1 veya SMARCA4 proteininin kusurlu bir gen nedeniyle hücrelerde eksik olup olmadığını belirlemek için tümör dokusunun özel olarak boyanması gerekebilir. Sitogenetik ve moleküler genetik, bu genetik kusuru doğrudan tespit etmek için kullanılabilmektedir.
Tümör dokusunda gerçekten bir SMARCB1 veya SMARCA4 mutasyonu mevcutsa, kan (veya diğer tümörsüz doku) genellikle bir germ hatti mutasyonunu, yani rabdoid tümör yatkınlık sendromunu (RTPS) ekarte etmek için moleküler genetik kullanılarak incelenir. Bu durumda, kan hücreleri (veya diğer doku hücreleri) de değişmiş geni içerir. Özellikle iki yaşın altındaki çocuklarda, vücudun farklı bölgelerinde tümörleri olan hastalarda (senkron tümörler) veya ailesel tümör öyküsü olduğunda, yani ailede tümör öyküsü varsa, germ hattı mutasyonundan şüphelenilir.
Doku örneği alınmadan önce rabdoid tümör yatkınlık sendromunun (RTPS1 veya RTPS2) varlığı biliniyorsa, bireysel vakalarda (örneğin çocuklar ameliyat edilemeyecek kadar hasta ise) sadece kan testi kullanılarak da tanı konulabilir. Bunun nedeni, kan hücrelerinde SMARCB1 veya SMARCA4 mutasyonu olan küçük bir çocukta görüntüleme yoluyla tespit edilen kötü huylu bir tümörün neredeyse kesinlikle bir AT/RT olmasıdır. Ancak kan testi doku örneğinin yerini alamaz.
Tümör dokusunun SMARCB1 veya SMARCA4 mutasyonu açısından incelenmesine ek olarak, DNA metilasyon profilinin belirlenmesi de giderek daha önemli hale gelmektedir. Bu moleküler genetik yöntem kullanılarak AT/RT üç farklı moleküler (epigenetik) alt tipe veya alt gruba ayrılabilir (ATRT-MYC, ATRT-SHH, ATRT-TYR), bunlardan bazıları büyüme davranışları açısından da farklılık gösterir (ayrıca bkz. bölüm “Tedavi planlaması”).
Bilinmesi önemli: Bir germ hattı mutasyonu tespit edilirse, bu sadece hastanın prognozu üzerinde değil, aynı zamanda muhtemelen aile üyeleri açısından da bir sonuç doğurmaktadır. Bu nedenle uygun önleyici tedbirler alınmalıdır (aşağıdaki rabdoid yatkınlık sendromu için önerilere bakınız).
Hastalığın yaygınlığını araştıran tetkiler (tümör evrelemesi)
Eğer rabdoid tumor tanısı kesinleşirse, hastalığın tüm vücuttaki yaygınlığını araştırmak için ek tetkikler gereklidir. Rabdoid tümörler sıklıkla metastaz oluşturduğundan ve örneğin merkezi sinir sisteminde (MSS) ve böbrekte aynı anda (senkron rabdoid tümör) olmak üzere başlangıçtan itibaren vücudun farklı bölgelerinde de ortaya çıkabildiğinden, bir tümör tespit edildiğinde her zaman tüm vücudun kapsamlı bir görüntüleme incelemesi yapılır.
Kural olarak, MSS içindeki metastazı dışlamak için beyin ve omuriliğin manyetik rezonans görüntülemesi (manyetik rezonans tomografisi, MRT) yapılır ve en az bir kez tüm vücut MRT'si de yapılır. Sonuncusu uzak metastazları ve/veya senkron rabdoid tümörleri tespit etmeye veya dışlamaya yarar. Buna ek olarak, genellikle ameliyattan sonra beyin omurilik sıvısı (BOS) tümör hücreleri açısından incelenir. Beyin omurilik sıvısını elde etmek için lomber omurga bölgesinde bir ponksiyon (lomber ponksiyon) yapılması gerekmektedir.
Tedavi başlanmadan yapılması gereken tetkikler
Tedaviye hazırlık olarak, örneğin elektrokardiyografi (EKG) ve/veya kalp ultrasonografisi (ekokardiyografi, EKO) ile kalp fonksiyonlarının kontrolü veya beyin dalgalarını incelemek için elektroensefalografi (EEG) gibi başka tetkikler de yapılabilir. Geniş çaplı kan tetkileri ile bazı organların fonksiyonları (örneğin böbrek ve karaciğer) değerlendirilir, tedavi öncesi ve tedavi sırasında dikkate alınması gereken metabolik bozukluklar tespit edilebilir. Tedavi sırasında meydana gelebilecek değişiklikler erken fark edilebilir ve bu tür ilk bulgular ve düzenli kontroller sayesinde daha etkili bir şekilde değerlendirilebilir.
Çocuğun genel durumu izin veriyorsa, tanı sırasında nöropsikolojik incelemeler (örneğin soru formları kullanılarak) de yapılabilir [bkz. nöropsikoloji]. Bunlar, çocuğun gelişim düzeyi ve bilişsel yeteneklerinin yanı sıra herhangi bir anormalliğin kayıt altına alınmasına ve daha sonra yapılacak araştırmalara temel teşkil etmesine yardımcı olur. Yaşam kalitesine ilişkin araştırmalar da eklenebilir.
Bilinmesi gereken nokta: Yukarıda sayılan tetkiklerim tümü her hasta için gerekli olmayabilir. Öte yandan muhtemelen yukarıda sayılmayan başka tetkikler de gerekli olabilir. Çocuğunuzla ilgili hangi muayenelerin planlandığını ve her bir muayenenin neden gerekli olduğunu doktorunuza veya tedavi ekibinin diğer üyelerine sorunuz.
Şüpheli rabdoid tümör eğilimi sendromu için öneriler
Bir hastada germ hatti mutasyonu ve dolayısıyla rabdoid tümör gelişimine kalıtsal yatkınlık (rabdoid tümör yatkınlık sendromu, RTPS) tespit edilirse, hastalığın bir ebeveyn tarafından çocuğa aktarılmış olması mümkündür. Ancak vakaların büyük çoğunluğunda bu değişikliğe yeni bir mutasyon neden olmaktadır.
Ebeveynlerden kalıtsal olarak geçmesi halinde, hastanın biyolojik kardeşlerinde de rabdoid tümör gelişme riski artacaktır (ayrıca bkz. bölüm "Sebepleri"). Böyle bir riski ortadan kaldırmak veya gerekirse kardeşler için erken teşhis önlemleri almak için, tedavi ekibi hasta çocuğun ebeveynlerine bir kan testi yaptırmalarını önerecektir. Bu test pozitif çıkarsa, yani ebeveynlerden birinde de germ hattı mutasyonu varsa, kardeşlere de kan testi yapılmalıdır. Testler insan genetiği konusunda uzmanlaşmış laboratuvarlar tarafından gerçekleştirilir. Uygun danışmanlık da tavsiye edilir.
RTPS'nin ailede görüldüğü biliniyorsa ve daha önce sağlıklı olan bir aile üyesinde germline mutasyon bulunursa, etkilenen çocuğun/çocukların doğumdan itibaren fiziksel/nörolojik muayeneler, manyetik rezonans tomografi ve ultrasonografi (baş, karın ve göğüs) yoluyla yakından izlenmesi önerilir.
Tedavi planlaması
Tanı konduktan sonra tedavi planlaması yapılır. Mümkün olduğunca kişiye özel ve hastaya uyarlanmış (risk adaptasyonlu) bir tedavinin gerçekleştirilebilmesi amacıyla, tedavi ekibi tedavi planını hazırlarken hastadaki prognozu (tedavi başarısı, sağ kalım) etkileyen belirli faktörleri (risk ve prognoz faktörlerini) dikkate alır.
Merkezi sinir sisteminin rabdoid tümörleri (AT/RT) için en önemli prognostik faktörlerden biri hastanın tanı anındaki yaşıdır. Yaş, tedavinin ne kadar yoğun olabileceğini belirler ve dolayısıyla hastanın hayatta kalma şansını etkiler. Örneğin, rabdoid tümörler için çok etkili bir tedavi olan radyoterapi, 3 yaşın altındaki çocuklar için yalnızca sınırlı ölçüde bir seçenektir ve genellikle 12 aylıktan küçük çocuklar için kesinlikle bir seçenek değildir. Diğer standart tedavi yöntemlerine (cerrahi ve kemoterapi gibi) yönelik tolerans da çok küçük çocuklarda sınırlıdır.
Bir diğer önemli prognostik faktör de AT/RT'nin moleküler alt tipidir, çünkü bu da hastalığın seyrini etkileyebilir (bkz. bölüm “Tanı / doku örneği alınması”). ATRT-TYR alt tipine sahip hastaların iyileşme şansı genellikle diğer iki alt tipten birine (ATRT-MYC, ATRT-SHH) sahip hastalardan daha yüksektir. Bu nedenle alt tipin bilinmesi, beklenen prognoz hakkında çıkarımlar yapılmasını sağlar.
Kalıtsal olup olmamanın yanı sıra rabdoid tümörün yeri ve boyutu da hastanın iyileşme şansını etkileyen diğer faktörlerden biridir. Kalıtsal bir hastalığın, yani rabdoid tümör yatkınlık sendromunun (RTPS) varlığı, tanı anında tümörün metastaz oluşturması olumsuz prognostik faktörler olarak kabul edilir. Her iki durumda da, tüm tümör belirtilerinin kalıcı olarak tamamen ortadan kaldırılması (olumlu bir prognozu destekler) daha zor hale gelir. Hastalığın kemoterapiye verdiği yanıt da önemli bir prognostik faktördür.
Tüm faktörler, her hasta için mümkün olan en iyi tedavi sonucunu elde etmek amacıyla tedavi planlamasında dikkate alınmaktadır.
Tedavi
Rabdoid tümörlü bir hastanın tedavisi mutlaka çocuk onkolojisi merkezi tarafından yapılmalıdır. Bu merkezlerde kanser hastası çocukların tedavisi konusunda en modern tedavi yöntemlerine alışık, eğitimli doktor ve sağlık ekibi bulunmaktadır. Bu bölümlerde çalışan doktorlar hastalarını, hastalık tiplerine özgü çalışma grupları ile yakın temas halinde, birlikte geliştirdikleri ve sürekli yenilenen tedavi planlarına göre tedavi ederler.
Tedavinin amacı yüksek oranda iyileşme sağlamanın yanında, tedaviye bağlı yan etki ve geç dönem etkileri olabildiğince en aza indirmektir. Bu durum, rabdoid tümörlü genellikle çok genç hastalar için büyük bir zorluk teşkil etmektedir. Bebekler ve küçük çocuklar özellikle hassas durumdadır; saldırgan tedavinin akut yan etkilerinden ve genellikle ciddi uzun vadeli sonuçlarından büyük ölçüde zarar görürler ve bu nedenle genellikle tedavi edilmeleri zordur. Bu nedenle tedaviden önce veya tedavi sırasında en önemli adım, bir tedavi girişiminin yapılıp yapılmayacağına veya devam ettirilip ettirilmeyeceğine ve eğer yapılacaksa bunun iyileşmeye mi (küratif tedavi) yoksa ağrının giderilmesine mi (palyatif tedavi) yönelik olacağına karar vermektir.
2007'den uluslararası SIOPE ATRT01 tedavi çalışmasının 2021 ortalarında açılışına kadar, rabdoid tümörlü hastalar Avrupa EU-RHAB veri tabanı çerçevesinde tek tip bir tedavi planına göre tedavi edilmiştir (konsensüs /uzlaşı tedavi stratejisi olarak adlandırılır, ayrıca bkz. bölüm “Tedavi iyileştirme araştırmaları ve veri bankası”). Aşağıda açıklanan tedavi uygulamaları bu uzlaşı tedavi stratejisine ve Pediatrik Onkoloji ve Hematoloji Derneği'nin (GPOH) AT/RT için güncel kılavuzlarına dayanmaktadır.
Tedavi yöntemleri
Rabdoid tümörü olan hastalar cerrahi (amelyat), kemoterapi ve radyoterapi ile tedavi edilebilir. Bazı durumlarda, yüksek doz kemoterapi ve ardından daha önce alınan kan kök hücrelerinin yeniden nakli (otolog kök hücre nakli) de bir seçenek olabilir. Hangi yöntemin hangi kombinasyonda kullanılacağı öncelikle hastanın yaşı ve sağlık durumunun yanı sıra tümörün türü, yeri ve boyutuna (ve dolayısıyla ameliyat edilebilirliğine) bağlıdır.
Prensip olarak, ameliyat ve radyoterapi rabdoid tümörü olan hastalar için en umut verici ve dolayısıyla en önemli tedavi yöntemleridir. Ancak bunlar tüm hastalara uygulanamaz. Örneğin radyoterapi sadece belirli bir yaştan itibaren mümkündür. Kemoterapi (ve muhtemelen yüksek doz kemoterapi) hayatta kalma şansını artırmaya yardımcı olabilir ve – özellikle çok küçük çocuklarda – radyoterapinin kullanılmasına kadar geçen süreyi uzatabilir veya onun yerini alabilir.
Ameliyat (cerrahi girişim, operasyon)
Rabdoid tümörlü bir hastanın tedavisinde ilk adım, tümörün ameliyat edilebilir olması koşuluyla, tümörü mümkün olduğunca tamamen almaya çalışmaktır. Çünkü çalışmalar, tümör ne kadar büyük ölçüde çıkarılabilirse hayatta kalma şansının o kadar yüksek olduğunu defalarca göstermiştir. Metastazı olmayan bölgesel bir tümör olması durumunda, tümörün tamamen çıkarılmasını sağlamak için cerrahi girişim tekrarlanabilir. Ancak ne yazık ki birçok hastada (yani hastaların yaklaşık %70'inde) çok fazla sağlıklı doku çıkarmadan ve hastanın yaşam kalitesini tehlikeye atmadan tümörü tamamen çıkarmak mümkün değildir. Bunun nedeni tümörün genellikle uygun olmayan yerleşimi, hastaların genç yaşta olması ve tanı anında tümörün metastazının [bkz. metastaz oluşturma sık görülmesidir.
Bir nöroşirürji (beyin cerrahisi) ameliyatı yapılırsa, sonraki kemoterapiye hazırlık olarak bir beyin ventrikülü (serebral ventrikül) bağlantısı da oluşturulur. İlaçlar bu ventrikül yolundan doğrudan beyin omurilik sıvısına (BOS) verilebilir (bu örneğin bir Ommaya rezervuarı veya bir Rickham rezervuarı olabilir) (sonraki bölüme bakınız).
Kemoterapi
Ameliyatın ardından hastanın iyileşme şansını artırmak için yoğun kemoterapi uygulanır. Kemoterapide kanser hücrelerinin büyümesini önleyip onları yok eden hücre büyümesini engelleyici ilaçlar (sitostatik ilaçlar) kullanılır. Tedavinin etkinliğini artırmak için çeşitli sitostatik ilaçlar farklı kombinasyonlarda kullanılır ve bloklar halinde uygulanır.
Mevcut kılavuzlara göre standart kemoterapi, bir indüksiyon (başlangıç, hücum) ve bir konsolidasyon (pekiştirme) aşaması olmak üzere iki aşamadan oluşur ve on iki kemoterapi kürüne kadar uygulanabilir. İndüksiyon tedavisi tümör ve/veya tümör hücresi sayısında mümkün olan maksimum kontrolü sağlamayı, konsolidasyon tedavisi ise elde edilen sonuçları korumayı amaçlar. Kullanılan ilaçlar arasında doksorubisin (DOX) ve ifosfamid, karboplatin ve etoposid (ICE olarak kısaltılır) veya vinkristin, siklofosfamid ve aktinomisin D (VCA) kombinasyonları yer alır ve bunlar dönüşümlü olarak intravenöz yoldan uygulanır.
Bu ilaçlar kan dolaşımı yoluyla tüm vücuda dağılır, bu nedenle aynı zamanda sistemik kemoterapi olarak da adlandırılır. Merkezi sinir sistemindeki tümör hücrelerine daha iyi ulaşabilmek için sitostatik ilaç metotreksat (MTX) beyin ventrikülü üzerinden doğrudan beyin omurilik sıvısına uygulanır. Bu intraventriküler veya intratekal kemoterapi önemlidir çünkü kan-beyin bariyeri ilaçların beyne sadece sınırlı bir ölçüde geçmesine izin verir.
Işın tedavisi (radyoterapi)
Hastanın tedavi sırasındaki yaşına bağlı olarak, ışın tedavisi (radyoterapi) sırasında veya sonrasında uygulanabilir. Işın tedavisinde cilt üzerinden etkilenen bölgeye yüksek enerjili, elektromanyetik ışınlar uygulanır. Işınlar tümör hücrelerinin bölünmesini engelleyerek, ölmelerini sağlar.
Tümörün tamamen cerrahi olarak çıkarılmasının yanı sıra radyoterapi, rabdoid tümörler için bugüne kadarki en önemli tedavi yöntemlerinden biridir. Bu yöntemle AT/RT tedavisinde büyük başarılar elde edilmektedir ve daha önce de gösterildiği gibi hastalar erken radyoterapiden fayda görmektedir. Ancak tedavinin geç yan etkileri nedeniyle kullanımı sınırlıdır. Bu durum özellikle beyinleri hala gelişmekte olan bebekler ve küçük çocuklar için geçerlidir: Gelişimin erken bir evresinde radyoterapi, diğer şeylerin yanı sıra, normal zeka gelişiminde (bilişsel gelişim) ciddi bozulmalara yol açabilir. Bu nedenle kemoterapi ve gerekirse yüksek doz kemoterapi, radyoterapiyi mümkün olduğunca geciktirmek veya tamamen kaçınmak için kullanılır.
Radyoterapi düşünülüyorsa, ışınlama zamanı, hedef hacim, radyasyon türü (fotonlar veya protonlar) ve radyasyon dozu hastanın yaşına, dokunun gelişimsel duyarlılığına ve hastalığın prognostik faktörlerine bağlıdır:
AT/RT için mevcut kılavuza göre, radyoterapi 12–18 aylıktan itibaren lokalize tümörü olan çocuklar için düşünülmektedir. Tümör bölgesi 54 Gray'e (Gy) kadar radyasyon dozu ile ışınlanır. Tümörü beyin omurilik sıvısı yoluyla merkezi sinir sistemine yayılmış çocuklarda (leptomeningial metastatik tümörler), üç yaşından itibaren beyin ve omurilik radyoterapisi (kraniospinal radyoterapi) düşünülür. Bu durumda, tüm merkezi sinir sistemine 35,2 Gy'lik bir radyasyon dozu artı primer tümöre ek bir doz (boost) (toplam 55 Gy'ye kadar) önerilir.
Yoğunluk ayarlı radyoterapi (IMRT) gibi modern radyoterapi teknikleri, sağlıklı dokuya radyasyon hasarının en aza indirilmesini sağlar [bkz. yoğunluk ayarlı radyoterapi]. Bazı hastalarda geleneksel radyoterapi (fotonlar kullanılarak) yerine proton tedavisi (proton ışınları kullanılarak) de düşünülebilir, örneğin çok küçük çocuklarda veya proton tedavisi geleneksel radyoterapiye oranla açıkça avantajlıysa. Bunun nedeni, proton tedavisinin özellikle doku dostu ve etkili olmasıdır.
SIOPE ATRT01 çalışması hakkında not: Bu çalışmanın amaçlarından biri, bebeklerde radyasyona bağlı geç hasarın, hastanın iyileşme şansını azaltmadan, radyoterapi olmaksızın yüksek doz kemoterapi kullanılarak önlenip önlenemeyeceğini araştırmaktır. Bu amaçla, yüksek doz kemoterapinin mevcut konvansiyonel radyokemoterapiden (12 kür kemoterapi artı radyoterapi) daha iyi olup olmadığı araştırılmaktadır. Çalışmaya metastazı olmayan ve rabdoid tümör eğilim sendromu (RTPS) bulunmayan 12–35 aylık çocuklar dahil edilecektir. Araştırma konusu ve tedavi süreci hakkında daha fazla bilgiyi “SIOPE ATRT 01 tedavi çalışması” hakkındaki ek bilgilerde bulabilirsiniz.
Yüksek doz kemoterapi ve otolog kök hücre nakli
Bazı hastalarda konsolidasyon aşamasında yukarıda açıklanan geleneksel kemoterapi yerine otolog kök hücre nakli ile birlikte yüksek doz kemoterapi düşünülebilir. Bu durumda, hasta altı kür standart kemoterapi alır (indüksiyon tedavisi, yukarıdaki "Kemoterapi" bölümüne bakınız), ardından karboplatin ve tiotepa (CARBO/TT) ile tedavi edilir. Böyle bir uygulamada dayanıklı tümör hücrelerini de yok edecek yoğunlukta yüksek dozda sitostatik ilaç verilir.
Ancak yoğun tedavi sadece kanser hücrelerini değil, kemik iliğindeki kan yapıcı (hematopoetik) sistemi de yok ettiğinden, hematopoetik kök hücreler (kan kök hücreleri) yüksek doz kemoterapiden önce hastanın kemik iliğinden veya kanından ayrıştırılır ve tedavi tamamlandıktan sonra geri aktarılır (nakledilir). Uzmanlar bunu otolog hematopoietik kök hücre nakli (otolog HSCT / otolog KHN veya otolog KIT veya SCT olarak kısaltılır) olarak da adlandırmaktadır.
Ancak bu tedavinin ön koşulu, kötü huylu hücrelerin büyük bir kısmının standart kemoterapi ile yok edilmiş, yani hastanın remisyona girmiş olmasıdır. Bu oldukça yoğun ve riskli bir tedavi olduğu için hastanın yaşı ve genel sağlık durumu da önemlidir.
Bugüne kadar, yüksek doz kemoterapinin tek başına standart kemoterapiden (12 aydan küçük çocuklarda) veya geleneksel kombine kemoterapi/radyoterapiden (12 ay ve üzeri çocuklarda) daha az etkili olup olmadığı net değildir. Avrupa çapında 1 Haziran 2021'den beri devam eden SIOPE ATRT01 şemsiye çalışması bu soruları yanıtlamayı amaçlamaktadır.
SIOPE ATRT01 çalışmasıyla ilgili not: Çalışma, 12–35 aylık çocuklarda indüksiyon kemoterapisini takiben konsolidasyon yöntemi olarak yüksek doz kemoterapinin (radyoterapi olmadan) standart kombine radyoterapi/kemoterapi (indüksiyon ve konsolidasyon kemoterapisi artı radyoterapi kombinasyonu) ile aynı derecede iyi sonuçlar verip vermediğini araştırmaktadır. 12 aylıktan küçük çocuklar veya radyoterapinin bir seçenek olmadığı çocuklar için, çalışma konsolidasyon aşamasında yüksek doz kemoterapinin geleneksel kemoterapiye devam etmekten daha iyi sağkalım oranlarına yol açıp açmadığını inceleyecektir.
Yeni tedavi yaklaşımları
Günümüzde mevcut olan yoğun tedavi yöntemlerine rağmen, AT/RT'li çocuklar için iyileşme beklentisi tatmin edici değildir. Bu durum özellikle yüksek riskli hastalar için geçerlidir (genç yaş, elverişsiz moleküler alt tip, germ hatti mutasyonu ve/veya metastatik tümör). Ayrıca, yoğun tedavi sadece akut yan etkilere değil, aynı zamanda uzun vadeli geç etkilere de yol açmaktadır (örneğin, ilgili gelişimsel bozuklukların eşlik ettiği hormon eksiklikleri veya işitme ve görme bozukluğu gibi duyusal bozukluklar). Bu durum hastanın yaşam kalitesi üzerinde kalıcı bir etkiye sahip olabilir.
Yeni ilaçlar ve tedavi yöntemleri bulmak amacıyla bilim insanları bu tümörlerle ilgili daha ileri araştırmalar üzerinde yoğun bir şekilde çalışmaktadır. Araştırmaların odağında rabdoid tümörlerin gelişmesine ve yayılmasına yol açan moleküler mekanizmalar yer almaktadır. Rabdoid tümörlerde değişime uğrayan sinyalizasyon basamaklarının araştırılmasıyla, rabdoid tümörlerin tedavisinde faydalı olabilecek çeşitli ilaçların tanımlanması mümkün olmuştur. Umut verici yeni tedavi yaklaşımları yapılacak çalışmalarda denenecektir.
AT/RT nüksü olan hastalar için kişiselleştirilmiş tedavi çalışmalarının bir parçası olarak çeşitli yaklaşımlar mevcuttur. Bunlar arasında desitabin veya ribociclib ilacı gibi epigenetik olarak aktif maddeler ve kontrol noktası inhibitörleri sayılabilir. Olası bir tedavi seçeneği de, örneğin MEMMAT protokolünün bir parçası olarak metronomik tedavidir.
Ne yazık ki, şu anda hasta çocuklar için sadece birkaç açık faz I/II çalışması bulunmaktadır. Örneğin NivEnt çalışması, yalnızca belirli bir moleküler ATRT alt tipine (ATRT-MYC) sahip çocukları içermektedir. EU-RHAB veri tabanının araştırmacıları yeni ilaçlar bulmak ve uygulamak için titizlikle çalışmaktadır. Bu her zaman klinik olarak kontrol edilen çalışmalar çerçevesinde gerçekleşmelidir (ayrıca bir sonraki bölüme bakınız).
Tedavi iyileştirme araştırmaları ve veri bankası
Büyük tedavi merkezlerinde rabdoid tümörü olan çocuk ve gençler, bu hastaların hayatta kalma şansını artırmayı amaçlayan kılavuzlara ve/veya tek tip (standartlaştırılmış) tedavi protokollerine göre tedavi edilmektedir. Bu tür tedavi protokollerine göre tedavi genellikle tedavi iyileştirme çalışmaları veya veri bankaları çerçevesinde yürütülür. Tedavi iyileştirme çalışmaları [tedavi iyileştirme araştırmaları] kontrollü klinik çalışmalardır. Bu çalışmaların amacı hastaları en yeni bilgiler ışığında tedavi etmenin yanında tedavi olanaklarını daha iyileştirmek ve geliştirmektir.
Tanı aldıkları sırada herhangi bir çalışma mevcut olmadığı veya mevcut bir çalışmanın dahil edilme kriterlerini karşılamadıkları için bir çalışmaya katılmayan hastalar bir veri bankasında kayıt altına alınırlar (register). Böyle bir veri bankasının (veri tabanının) birincil amacı, tümör biyolojisinin daha iyi anlaşılmasını sağlayacağı umulan tüm klinik, moleküler genetik ve tedaviyle ilgili hasta verilerini kaydetmektir. Buna ek olarak, sorumlu veri tabanı kayıt merkezi genellikle tedavi ekibini o anda bilinen mümkün olan en iyi tedaviye dayalı (bağlayıcı olmayan) tedavi önerileriyle destekler, böylece hasta bir çalışma dışında da en uygun tedaviyi alır.
Şu anda AT/RT hastaları için SIOPE ATRT 01 çalışması ve EU-RHAB kayıtları mevcuttur. Diğer çalışmalar planlama aşamasındadır. Mevcut çalışmalar / veri tabanları hakkında daha fazla bilgiyi aşağıda bulabilirsiniz.
Avrupa Rabdoid Kayıtları (EU-RHAB-Register)
Rabdoid tümörler çok seyrek görülen tümörler olduğundan, 2007 yılında İtalya'da düzenlenen bir uzlaşma konferansında – GPOH uzmanlık derneğinden araştırmacıların girişimiyle – Avrupa'nın bir bölümündeki tüm rabdoid tümörlü hastaların Avrupa Rabdoid Kayıt Merkezine (EU-RHAB Kayıt Merkezi, EU-RHAB veri tabanı) kaydedilmesine ve standart bir tedavi yöntemine (konsensüs tedavisi) göre tedavi edilmesine karar verilmiştir. Tedavi rejimi, uluslararası bir uzmanlar ağının parçası olarak geliştirilmiştir ve “Tedavi” bölümünde ve AT/RT için güncel kılavuzda açıklanmıştır. Hasta bir çalışmanın deneme kolunda tedavi edilmediği sürece, tüm rabdoid tümörler için (vücuttaki konumlarından bağımsız olarak) standarttır ve bireysel hastaya göre uyarlanmalıdır.
Dünya çapında çok sayıda ülke hastalarını bu EU-RHAB veri tabanına bildirmektedir. Almanya'da kanserli çocuk ve gençleri tedavi eden hastaneler, ilgili hastaları uzman kuruluşun ilgili çalışmalarına veya veri abanlarına bildirmekle yasal olarak yükümlüdürler. Tedavinin veri tabanının önerisine uygun olarak yürütülüp yürütülmeyeceğine ilişkin karar her bir kliniğe aittir. Ancak ebeveynler olarak çocuğunuzun EU-RHAB önerilerine uygun olarak tedavi edilip edilemeyeceğini sorma hakkına sahipsiniz. Avrupa kayıt merkezi, Augsburg Üniversite Hastanesi Çocuk Hastanesi'nde Profesör Dr. Dr. med. Michael C. Frühwald yönetiminde bulunmaktadır. Not: Hastaların veri tabanında kayıt altına alınmasına 24.04.2023 tarihinden itibaren geçici olarak ara verilmiştir. Kısa süre içinde yeni bir veri tabanı (EU-RHAB 2.0) açılacaktır.
Tedavi çalışması SIOPE ATRT 01
Temmuz 2021'de, AB RHAB veri tabanının bir parçası olarak geliştirilen AT/RT'li çocuklar için uluslararası, çok merkezli bir tedavi çalışması başlatıldı: SIOPE ATRT01 “şemsiye” çalışması. Bu bağlamda “şemsiye” terimi, çalışmanın birkaç bölümden oluştuğu ve etkilenen hastaları risk profillerine bağlı olarak bir tedavi koluna atadığı anlamına gelir.
0–17 yaş arası Avrupa’daki bütün hastalar Umbrella çalışmasına dahil edilecektir. Bütünleştirilmiş randomize (rastgele) tedavi iyileştirme çalışması 12–35 aylık hastalar için tasarlanmıştır [bkz. randomizasyon]. Çalışmaya Almanya'dan ve neredeyse tüm Avrupa ülkelerinden (Fransa, İsviçre, İngiltere, İspanya ve Portekiz dahil) çok sayıda tedavi merkezi katılmaktadır. Çalışma merkezi Augsburg Üniversite Hastanesi'nde Prof. Dr. med. Michael C. Frühwald yönetiminde bulunmaktadır.
Yeni çalışmalar
EU-RHAB'ın bir parçası olarak, toplanan veriler temel alınarak yeni çalışmalar da geliştirilmektedir. Şu anda rabdoid tümörlere karşı etkili olması muhtemel yeni bir bileşenin test edilmesi için bir faz I/II çalışması planlanmaktadır. Ayrıca, nükseden hastalığı olan hastaların tedavisi için yeni bir tedavi protokolü geliştirilmektedir.
Tedavi başarısı (prognoz)
Merkezi sinir sisteminin rabdoid tümörü (AT/RT) olan çocukların iyileşme şansı (prognoz), EU-RHAB çerçevesinde standartlaştırılmış, multimodal tedavi stratejileri sayesinde önemli ölçüde artmıştır. En elverişli tedavi grubunda 5 yıllık sağkalım oranları artık %71,5'in üzerindedir. Ancak olumsuz prognostik faktörlere sahip hastalar için, yoğun tedaviye rağmen sağkalım beklentileri hala kötüdür. EU-RHAB kayıtlarının sonuçlarına göre ortalama olarak (yani tüm risk gruplarında) 5 yıllık sağkalım oranı %35-40 civarındadır.
Belirleyici prognostik faktörler özellikle hastanın tanı anındaki yaşı, tümör tipi (kalıtsal veya kalıtsal olmayan, moleküler alt tip) ve tümörün boyutu, yeri ve yayılımı ve dolayısıyla tümörün tamamen çıkarılma olasılığıdır (ayrıca bkz. “Tedavi planlaması” bölümü). Hayatta kalma şansı buna dayanarak hastadan hastaya değişir:
- Lokal, metastatik olmayan, kolayca ameliyat edilebilen ve kalıtsal olmayan rabdoid tümörü olan ve tanı anında 3 yaşından büyük olan hastaların, tümörün ameliyat sırasında tamamen çıkarılabilmesi ve erken radyoterapinin mümkün olması koşuluyla, iyileşme beklentileri genellikle iyidir.
- Aynı durumda olan ancak yaşı 1 ile 3 arasında olan çocuklarda nüks riski daha yüksektir ve dolayısıyla iyileşme olasılığı daha düşüktür.
- Prognoz, özellikle AT/RT'nin belirli moleküler alt tipleri de mevcutsa, 1 yaşın altındaki bebekler ve küçük çocuklar için özellikle kötüdür.
- Aynı durum, olumsuz prognostik faktörlere, yani yüksek riskli rabdoid tümöre sahip diğer tüm hastalar için de geçerlidir. Bunlar arasında germ hatti mutasyonu, yani rabdoid tümör geliştirmeye yatkınlığı olan hastaların yanı sıra ameliyat edilemeyen primer tümörü veya metastatik hastalığı olan hastalar da yer almaktadır.
Bununla birlikte, olumsuz prognoz faktörlerine rağmen tedaviden (cerrahi, kemoterapi, muhtemelen yüksek doz kemoterapi ve radyoterapi) fayda gören hastalar da vardır, dolayısıyla uzun süreli sağkalım mümkündür. Bu yüksek riskli hastalar için de iyileşme şansını artırmak amacıyla tedavi çalışmalarının bir parçası olarak yeni moleküler tedavi yaklaşımları araştırılmaktadır.
Uyarı: Yukarıda sözü edilen sağkalım oranları istatistiksel verilerdir. Yalnızca tüm rabdoid tümörlü hastaları için geçerli ve gerçeğe uygun bir ifade oluşturmaktadırlar. Bir hastanın iyileşeceği veya iyileşmeyeceği konusunda istatistiğe dayanarak bir şey söylemek mümkün değildir.
 Rabdoid tümörleri (AT/RT) - Kısa Bilgiler (323KB)
Rabdoid tümörleri (AT/RT) - Kısa Bilgiler (323KB)Kaynakça 
- Frühwald MC et al.: S1-Leitlinie „Atypische Teratoide / Rhabdoide Tumoren“ der Gesellschaft für Pädiatrische Onkologie und Hämatologie. Registernummer 025-037 AWMF online 2024 [URI: https://register.awmf.org/ assets/ guidelines/ 025-037l_S1_Atypische-Teratoide-Rhabdoide-Tumoren_2024-05.pdf]
- Rutkowski S, Pfister S, Frühwald M, Fleischhack G, Korinthenberg R, Bison B, Hahn G, Mentzel H-J, Langen K-J, Hernáiz-Driever P, Pietsch T: Leitsymptome und Diagnostik der ZNS-Tumoren im Kindes- und Jugendalter. Gemeinsame Leitlinie der Gesellschaft für Neuropädiatrie und der Gesellschaft für Pädiatrische Onkologie und Hämatologie AWMF online, 2024 [URI: https://register.awmf.org/ assets/ guidelines/ 025-022l_S1_Leitsymptome-Diagnostik-ZNS-Tumoren-Kinder-Jugendliche_2024-06.pdf]
- Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, Soffietti R, von Deimling A, Ellison DW: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro-oncology 2021 Aug 2; 23: 1231 [PMID: 34185076]
- Frühwald MC, Nemes K, Boztug H, Cornips MCA, Evans DG, Farah R, Glentis S, Jorgensen M, Katsibardi K, Hirsch S, Jahnukainen K, Kventsel I, Kerl K, Kratz CP, Pajtler KW, Kordes U, Ridola V, Stutz E, Bourdeaut F: Current recommendations for clinical surveillance and genetic testing in rhabdoid tumor predisposition: a report from the SIOPE Host Genome Working Group. Familial cancer 2021,online ahead of print [PMID: 33532948]
- Erdmann F, Kaatsch P, Grabow D, Spix C: German Childhood Cancer Registry - Annual Report 2019 (1980-2018). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2020 [URI: https://www.kinderkrebsregister.de/ typo3temp/ secure_downloads/ 42507/ 0/ 1c5976c2ab8af5b6b388149df7182582a4cd6a39/ Buch_DKKR_Jahresbericht_2019_komplett.pdf]
- Frühwald MC, Hasselblatt M, Nemes K, Bens S, Steinbügl M, Johann PD, Kerl K, Hauser P, Quiroga E, Solano-Paez P, Biassoni V, Gil-da-Costa MJ, Perek-Polnik M, van de Wetering M, Sumerauer D, Pears J, Stabell N, Holm S, Hengartner H, Gerber NU, Grotzer M, Boos J, Ebinger M, Tippelt S, Paulus W, Furtwängler R, Hernáiz-Driever P, Reinhard H, Rutkowski S, Schlegel PG, Schmid I, Kortmann RD, Timmermann B, Warmuth-Metz M, Kordes U, Gerss J, Nysom K, Schneppenheim R, Siebert R, Kool M, Graf N: Age and DNA-methylation subgroup as potential independent risk factors for treatment stratification in children with Atypical Teratoid/Rhabdoid Tumors (ATRT). Neuro-oncology 2020, [PMID: 31883020]
- Kuhlen M, Wieczorek D, Siebert R, Frühwald MC: How I approach hereditary cancer predisposition in a child with cancer. Pediatric blood & cancer 2019, 66:e27916 [PMID: 31342632]
- Frühwald MC, Furtwängler R: Das Europäische Rhabdoidregister – Basis für klinischen Fortschritt in der Behandlung einer sehr seltenen Tumorerkrankung. WIR - die Zeitschrift der Deutschen Leukämie-Forschungshilfe e.V. und der Deutschen Kinderkrebsstiftung 2018, 2/18
- Nemes K, Clément N, Kachanov D, Bens S, Hasselblatt M, Timmermann B, Schneppenheim R, Gerss J, Siebert R, Furtwängler R, Bourdeaut F, Frühwald MC, EU-RHAB consortium: The extraordinary challenge of treating patients with congenital rhabdoid tumors-a collaborative European effort. Pediatric blood & cancer 2018, 65:e26999 [PMID: 29418059]
- Nemes K, Frühwald MC: Emerging therapeutic targets for the treatment of malignant rhabdoid tumors. Expert opinion on therapeutic targets 2018, 22: 365 [PMID: 29528755]
- Frühwald MC, Hasselblatt M: Rhabdoide Tumoren des ZNS, der Nieren und des Weichteilgewebes. in: Niemeyer C, Eggert A (Hrsg.): Pädiatrische Hämatologie und Onkologie, Springer-Verlag GmbH Deutschland 2. vollständig überarbeitete Auflage 2018, 402 [ISBN: 978-3-662-43685-1]
- Nemes K, Bens S, Bourdeaut F, Hasselblatt M, Kool M, Johann P, Kordes U, Schneppenheim R, Siebert R, Frühwald MC, In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds): Rhabdoid Tumor Predisposition Syndrome. GeneReviews 2017 [PMID: 29215836]
- Bartelheim K, Nemes K, Seeringer A, Kerl K, Buechner J, Boos J, Graf N, Dürken M, Gerss J, Hasselblatt M, Kortmann RD, Teichert von Luettichau I, Nagel I, Nygaard R, Oyen F, Quiroga E, Schlegel PG, Schmid I, Schneppenheim R, Siebert R, Solano-Paez P, Timmermann B, Warmuth-Metz M, Frühwald MC: Improved 6-year overall survival in AT/RT - results of the registry study Rhabdoid 2007. Cancer medicine 2016, [PMID: 27228363]
- Frühwald MC, Biegel JA, Bourdeaut F, Roberts CW, Chi SN: Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro-oncology 2016, 18: 764 [PMID: 26755072]
- Benesch M, Bartelheim K, Fleischhack G, Gruhn B, Schlegel PG, Witt O, Stachel KD, Hauch H, Urban C, Quehenberger F, Massimino M, Pietsch T, Hasselblatt M, Giangaspero F, Kordes U, Schneppenheim R, Hauser P, Klingebiel T, Frühwald MC: High-dose chemotherapy (HDCT) with auto-SCT in children with atypical teratoid/rhabdoid tumors (AT/RT): a report from the European Rhabdoid Registry (EU-RHAB). Bone marrow transplantation 2014, 49: 370 [PMID: 24419520]